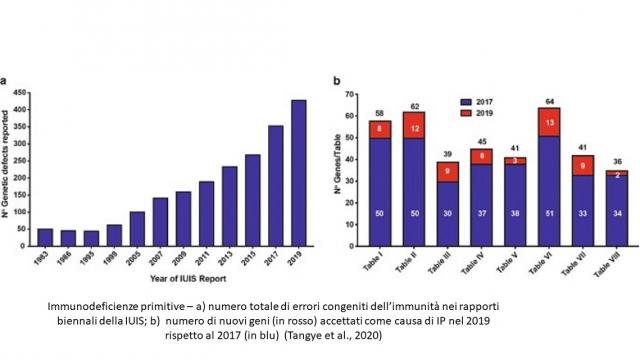
Crescita del numero di geni mutati e delle IP da essi provocati nel corso degli anni. Da Tangye et al [15]
28/08/2020
Immunodeficienze primitive - Introduzione e classificazione
(Ultimo aggiornamento: 25/01/2021)
Introduzione
Il termine di immunodeficienze primitive (IP), indica un gruppo eterogeneo di difetti che causano anomalo sviluppo di uno o più tipi cellulari del sistema immunitario (linfociti B e T, cellule natural killer, cellule dendritiche, fagociti) o alterata produzione delle citochine e del complemento che sono necessari per il buon funzionamento del sistema immunitario nativo e acquisito [1]. Le IP quasi sempre sono la conseguenza di mutazioni genetiche ereditabili, ma sono note varianti acquisite, per esempio i rari casi di autoanticorpi anti-interferone gamma osservati nel 95% circa pazienti con infezioni disseminate da micobatteri atipici [1] Le IP si manifestano con una predisposizione a infezioni, allergie, malattie autoimmuni e auntoinfiammatorie, neoplasie [2–4].
Le IP sono quasi tutte causate da mutazioni monogeniche germline che possono causare perdita dell’espressione o perdita della funzione (LOF, mutazioni amorfiche o ipomorfiche) o guadagno di funzione (gain of function, GOF, mutazioni ipermorfiche) della proteina codificata dal gene mutato [5]. Mutazioni eterozigoti possono manifestarsi come trait autosomico dominante mediante GOF, aploinsufficienza o dominanza negativa. Le mutazioni bialleliche causano tratti AR per LOF (raramente GOF) della proteina, mentre nel caso dei trait X-linked sono interessati geni del cromosoma X in stato emizigote nei maschi o omozigote nelle femmine [5]. Le proteine mutate in condizioni normali svolgono in genere funzioni fondamentali per lo sviluppo, la maturazione e il funzionamento dei linfociti, dei fagociti o di cellule extra emopoietiche importanti per il normale funzionamento del sistema immunitario. A causa delle mutazioni è alterata la risposta del sistema immunitario agli antigeni estranei (malattie infettive), agli autoantigeni (malattie autoimmuni, neoplasie) o altri agenti esterni [2,4,6,7].
La precocità della diagnosi e dell’inizio della terapia è fondamentale per migliorare la prognosi e la qualità dei vita dei pazienti [6]. Purtroppo, questo è un obiettivo lontano dall’essere raggiunto, se è vero che l’intervallo fra l’inizio dei sintomi e il riconoscimento dell’IP è stato anche di oltre vent'anni, in genere successivamente a numerosi episodi infettivi o quando si sono già instaurati danni d’organo irreversibili [8]. Le infezioni ricorrenti, specie quelle polmonari, le sequele di tali infezioni e le malattie neoplastiche cui i pazienti con IP sono predisposti sono causa di notevole aumento della morbilità e mortalità [6,9]. Nonostante sia oggi possibile una diagnosi molecolare delle varie IP, è necessario che i medici mantengano un alto indice di sospetto per queste malattie, soprattutto i pediatri e i medici di famiglia che, con molte probabilità, sono i primi ad incontrare la maggioranza dei pazienti con IP. Le infezioni ricorrenti sono la manifestazione d’esordio di molte IP e il loro numero, il tipo, la sede e le modalità di ricorrenza possono indirizzare verso uno dei principali raggruppamenti fenotipici nei quali le IP sono classificate, indirizzando verso la corretta diagnosi e guidando la scelta dei test diagnostici che, come vedremo, sono spesso di difficile ed elaborata esecuzione e non sono alla portata di tutti i laboratori, nemmeno nei paesi economicamente più floridi [10] .
Le IP sono classificate fra le malattie rare, sono esenti dal pagamento del ticket e sono regolate dalle norme previste dal decreto ministeriale 18 maggio 2001, n. 279 “Regolamento di istituzione della Rete nazionale delle malattie rare e di esenzione dalla partecipazione al costo delle relative prestazioni sanitarie ai sensi dell’articolo 5, comma 1, lettera b,) del decreto legislativo 29 aprile 1998 n.124” (S.O. alla G.U. n. 160 del 12 luglio 2001). La definizione di malattia rara non è univoca a livello internazionale. In Europa si definisce rara una malattia la cui prevalenza inferiore è inferiore a 5 casi ogni 10.000 abitanti [11] (Vedere " Immunodeficienze primitive - Epidemiologia")
Anche se le IP sono considerate rare, gli enormi progressi compiuti nella loro identificazione e terapia, hanno consentito di migliorare la comprensione dei meccanismi patologici e la terapia di numerose altre malattie più frequenti. Un esempio è la BTK, le cui mutazioni congenite sono la causa della agammaglobulinemia di Bruton. La scoperta di mutazioni somatiche (acquisite) di BTK nella leucemia linfatica cronica, e in alte neoplasie linfoidi a cellule B, ha consentito di sintetizzare gli inibitori della BTK, che si stanno dimostrando tra i farmaci più efficaci nella terapia di queste neoplasie [12]. (Vedere anche " Leucemia linfatica cronica - Fattori di rischio per la progressione a sindrome di Richter") Altri esempi sono la costruzione in laboratorio di farmaci mirati (targettati) in grado di inibire alcune mutazioni responsabili di IP [13]. Per esempio, gli inibitori di JAK sono usati nella terapia di disordini immunitari causati da mutazioni GOF di JAK1, STAT1 o STAT3 . Gli inibitori di PI3Kp110delta sono stati usati per il trattamento di pazienti con mutazioni GOF di PIK3D o mutazioni LOF di PIK3R1 [5]. E in letteratura vi sono altri esempi di terapie molecolari mirate che si sono rivelate utili nel trattamento di alcune IP [5] .
Classificazione delle immunodeficienze primitive
Non esiste un solo sistema di classificazione dell’eterogeneo gruppo di immunodeficienze primitive in grado di soddisfare le esigenze dei clinici, dei ricercatori e degli studenti [14]. La maggior parte dei clinici utilizza una classificazione funzionale, proposta dalla IULS (International Union of Immunology Societies) [15], che distingue le immunodeficienze primitive in gruppi in base al meccanismo immunologico la cui perturbazione è responsabile delle principali manifestazioni cliniche di laboratorio per quel gruppo di condizioni o sindromi . Da un punto di vista prevalentemente pratico, per esempio, possiamo distinguere fra immunodeficienze di deficit anticorpali, immunodeficienze combinate (che interessano contemporaneamente sia l’immunità cellulare che quella anticorpale), deficit del complemento, deficit dei fagociti eccetera. Tuttavia, questa suddivisione prevalentemente descrittiva in categorie funzionali presenta numerosi punti di sovrapposizione dal momento che i fagociti e il complemento, per esempio, sono considerati componenti dell’immunità innata ma in genere sono considerate separatamente per scopi prevalentemente didattici.
Nell’ultimo aggiornamento della IUIS sono riportati oltre 430 distinti errori congeniti dell’immunità o difetti genetici associati con uno stato di immunodeficienza [15] . Ed è probabile che la lista sia destinata ad allungarsi in futuro, dal momento che il comitato di esperti nel suo report biennale inserisce soltanto i difetti genetici per i quai esistono prove convincenti di patogenicità giudicate con criteri molto rigidi. E come per ogni classificazione, l’assegnazione di una condizione ad una categoria è spesso arbitraria. Per esempio, il deficit di ligando del CD40 (CD40L) è classificato sia fra i deficit anticorpali che fra le immunodeficienze combinate. Inoltre, questo sistema di classificazione potrebbe essere considerato scarsamente utile in alcuni contesti, per esempio nella pratica clinica o ambulatoriale. La Figura 1, ripresa dal riferimento bibliografico [15], riporta la crescita nel corso degli anni del numero di geni le cui mutazioni sono causa di IP e delle sindromi associati a tali mutazioni
Le immunodeficienze primitive sono raggruppate in 9 tabelle per un totale di 430 malattie e 404 difetti genetici noti come causa di IP, alle quali è stata aggiunta una decima tabella sulle fenocopie di errori congeniti dell’immunità;
- Immunodeficienze combinate
- Immunodeficienza combinate con caratteristiche sindromiche
- Deficit prevalentemente anticorpali
- Malattie da disregolazione immunitaria
- Difetti congeniti dei fagociti
- Difetti dell’immunità intrinseca e innata
- Malattia autoinfiammatorie
- Deficit del complemento
- Insufficienze congenite del midollo emopoietico
- Fenocopie degli errori congeniti dell’immunità
Il numero delle malattie considerate nelal classificazione IUILS [15] è superiore a quello dei difetti genetici, in quanto un difetto può provocare più di un fenotipo. E nella classificazione IULS vi sono 35 geni che sono presenti in più tabelle, a dimostrazione della complessità dei meccanismi che controllano le risposte immunitarie [15]. Al contrario, vi sono anche esempi di un fenotipo associato a genotipi differenti. Per esempio, le mutazioni bi-alleliche di ZNF341 [16], di IL6ST (che codifica per la gp130, un componente condivisa dai recettori per IL-6, IL-11, IL-27, LIF, OSM, CNTF), o di IL6R [17] sono tutte condizioni che assomigliano alla sindrome da iper IgE autosomica dominante causata da mutazioni dominanti negative di STAT3 [18]. Lo studio approfondito di questi pazienti ha permesso di scoprire nuovi meccanismi di regolazione del segnale di STAT3 (via il fattore di trascrizione ZNF341) e di definire nei dettagli le conseguenze dell’interruzione della via IL-6/IL-6R/gp130 e probabilmente di quella IL-11/IL-11R/gp130.(Vedere anche " Le sindromi da iper IgE - Introduzione" e "Le sindromi da iper IgE - Deficit di ZNF341 " e "Le sindromi da iper IgE - Deficit di IL6-R (HIES-5)" e "Le sindromi da iper IgE - Deficit di IL6ST (HIES-4) " e "Le sindromi da iper IgE - Deficit di STAT3 (sindrome di Job) ")
È stato calcolato che il genoma umano conterrebbe circa 2000 geni che intervengono nella risposta immunitaria [19]. Di questi, 1540 interverrebbero nella risposta immunitaria innata e 515 sarebbero associati con le risposte immunitarie a trattative o acquisite, compresi 200 geni che sono associati con entrambi i tipi di risposta (1-3). Molti di questi geni e loro mutazioni sono state scoperte dopo l’applicazione delle metodiche più sofisticate di sequenziamento dei geni, come la next generation DNA sequencing (NGS) (sequenziamento del DNA massivo in parallelo) che permettono di decifrare la sequenza dell’intero genoma o di sue porzioni in tempi relativamente brevi e a costi relativamente ridotti.
Caratteristiche principali delle immunodeficienze primitive
Nel proseguo della nostra trattazione ci soffermeremo brevemente sulle principali caratteristiche cliniche di ogni gruppo di IP, rimandando alle rispettive sezioni per la discussione dettagliata delle principali entità di ogni categoria. Infine, ci soffermeremo brevemente su un gruppo di sindromi genetiche in cui lo stato di immunodeficienza è parte di uno spettro di manifestazioni e malformazioni congenite ben caratterizzate le quali, generalmente, prevalgono sull’immunodeficienza stessa.
(Vedere anche " Immunodeficienze primitive - Cenni sulla risposta immunitaria" e "Immunodeficienze primitive - Introduzione e classificazione ")
1. Immunodeficienze combinate
Le immunodeficienze combinate (CID) sono delle rare sindromi da errori congeniti dell’immunità caratterizzate da gravissimi deficit quantitativi e/o qualitativi dei linfociti T con o senza difetti dei linfociti B (Tangye, Al-Herz et al. 2020). I pazienti presentano infatti gravi infezioni virali e fungine disseminate ad esito fatale se i bambini affetti non sono trattati in modo adeguato e con la dovuta tempestività [14]. Molte entità di questo gruppo sono oggi efficacemente risolvibili con il trapianto di midollo e, in qualche caso, con la terapia genica [20]. È importante però che i bambini affetti giungano al trapianto nelle migliori condizioni possibili, per cui è imperativo stabilire la diagnosi nel più breve tempo possibile e affidare i piccoli pazienti a centri con la adeguata esperienza nella diagnosi e trattamento delle IP [21]. Globalmente questo gruppo costituisce il 30-50% circa delle immunodeficienze congenite. (Vedere "Immunodeficienze primitive combinate - Classificazione ")
Le forme più gravi delle CID sono le SCID, la cui caratteristica fondamentale è l’assenza di linfociti T. Le SCID a loro volta sono suddivise in due sottotipi in base alla presenza (T-B+) o assenza (T-B-) dei linfociti B. Anche il numero delle cellule NK può essere informativo sul tipo di difetto genetico [1] . Le altre CID sono raggruppate nell’ambito delle “CID con decorso in genere meno grave delle SCID”. (Vedere "Le sindromi da iper-IgM ")
2.Immunodeficienza combinate con caratteristiche sindromiche
In questa categoria alquanto eterogenea sono comprese 9 sottogruppi di disordini caratterizzati dalla coesistenza di peculiari difetti congeniti di altri organi e apparati diversi da quello immunitario: a) CID con piastrinopenia congenita; b) CID da difetti di riparazione del DNA diversi da analoghi difetti che causano SCID; CID anomalie del timo con altre anomalie congenite; CID con displasia immunoossea; sindromi da iper-IgE; CID da difetti del metabolismo della vitamina B12 e dei folati; Displasia anidrotica ectodermica con immunodeficienza; CID da difetti dei canali del calcio; altri difetti immunitari.
- CID con piastrinopenia congenita (Sindrome di Wiskott-Aldrich)
-
Deficit prevalentemente anticorpali
In questa categoria sono compresi le immunodeficienze con defici prevalente della sintesi degli anticorpi. I deficit congeniti dell’immunità umorale sono come gruppo le IP più frequenti. Di questa categoria fanno parte: l’agammaglobulinemia di Bruton; il deficit grave di almeno due classi di immunoglobuline sieriche, fenotipo CVID; la grave riduzione delle IgG e IgA con IgM normali o aumentate, iper-IgM; i deficit dell’isotipo, catene leggere o funzionali con numero di linfociti B generalmente normale.
-
Malattie da disregolazione immunitaria
Questa categoria comprende sette sottogruppi di sindromi accomunate dalla perdita della tolleranza immunitaria che si manifesta con malattie autoimmuni e altre anomalie; le infezioni ricorrenti sono meno frequenti rispetto alle precedenti categorie. I sottogruppi sono: sindromi familiari con linfoistiocitosi emofagocitica ¸ sindromi familiari con linfoistiocitosi emofagocitica con ipopigmentazione; Difetti dei linfociti Tregolatori (Treg); sindromi linfoproliferative autoimmuni; disregolazione immune con colite; predisposizione alle infezioni da EBV e malattie linfoproliferative;
-
Difetti congeniti numerici e/o qualitativi dei fagociti
I fagociti sono cellule deputate all’ingestione ed eliminazione di agenti estranei. Le cellule fagocitarie principali sono i neutrofili e i monociti, questa categoria riunisce in quattro sottogruppi le anomalie primitive del numero e/o della funzione dei fagociti: neutropenie congenite; difetti della motilità; difetti del metabolismo ossidativo; altri difetti non linfoidi. (Vedere anache "DIfetti congeniti del numero e della funzione dei fagociti " )
-
Difetti dell’immunità intrinseca e innata
Difetti delle cellule NK, dei Toll-like Receptors (TLR) presentano infezioni virali ricorrenti e aumentato rischio di neoplasie. Si distinguono otto sottogruppi in questa categoria: predisposizione mendeliana alle infezioni micobatteriche; epidermodisplasia verruciforme; predisposizione a gravi infezioni virali; encefalite da Herpes simplex; predisposizione a gravi infezioni fungine ; predisposizione alla candidosi mucocutanea; deficit fei TLR con predisposizione alle infezioni batteriche; altri errori congeniti dell’immunità dipendenti da tessuti non emopoietici
-
Malattie autoinfiammatorie
Sono un gruppo di malattie derivanti da un’eccessiva liberazione di citochine proinfiammatorie che attivano diverse vie di segnalazione che causano febbre e danni sistemici ricorrenti. Questa categoria comprende tre sottogruppi: interferonopatie di tipo 1; difetti dell’infiammasoma; condizioni non correlate con l’infiammasoma
-
Deficit del complemento
Deficit delle proteine (C1.C4) che intervengono nelle prime tappe dell’attivazione del complemento si presentano con LES e infezioni ricorrenti da batteri capsulati: le infezioni ricorrenti da Neisseria meningitidis sono invece la caratteristica saliente dei deficit delle proteine C5-C9 e de componenti la via alternativa di attivazione del complemento.
-
Insufficienze congenite del midollo emopoietico
In questa categoria sono compresi alcuni errori genetici che causano un’insufficiente produzione di uno o più elementi cellulari del sangue periferico. La manifestazione iniziale è spesso un’anemia macrocitica, ma col passare degli anni si instaura una pancitopenia. L’anemia di Fanconi e la discheratosi congenita sono i due sottogruppi compresi in questa categoria dalla classificazione IUIS.(Vedere anche " Le Neutropenie - Classificazione e cause" )
-
Fenocopie degli errori congeniti dell’immunità
Le fenocopie degli errori congeniti dell’immunità sono oe sindromi che simulano un’immunodeficienza primitiva. Sono causate da mutazioni somatiche e non germline o da autoanticorpi, i casi piu rappresentativi sono le mutazioni dei recettori FAS che causano le sindromi linfoproliferative autoimmuni, gli autoanticorpi anti-IFNgamma e l’angioedema acquisito con autoanticorpi anti-inibitore di C1.
Bibliografia
[1] McCusker C, Upton J, Warrington R. Primary immunodeficiency. Allergy Asthma Clin Immunol 2018;14:61. https://doi.org/10.1186/s13223-018-0290-5.
[2] Kebudi R, Kiykim A, Sahin MK. Primary Immunodeficiency and Cancer in Children; A Review of the Literature. Curr Pediatr Rev 2019;15:245–50. https://doi.org/10.2174/1573396315666190917154058.
[3] Chi C-Y, Lin C-H, Ho M-W, Ding J-Y, Huang W-C, Shih H-P, et al. Clinical manifestations, course, and outcome of patients with neutralizing anti-interferon-γ autoantibodies and disseminated nontuberculous mycobacterial infections. Medicine (Baltimore) 2016;95. https://doi.org/10.1097/MD.0000000000003927.
[4] Amaya-Uribe L, Rojas M, Azizi G, Anaya J-M, Gershwin ME. Primary immunodeficiency and autoimmunity: A comprehensive review. J Autoimmun 2019;99:52–72. https://doi.org/10.1016/j.jaut.2019.01.011.
[5] Notarangelo LD, Bacchetta R, Casanova J-L, Su HC. Human inborn errors of immunity: An expanding universe. Science Immunology 2020;5. https://doi.org/10.1126/sciimmunol.abb1662.
[6] Jesenak M, Banovcin P, Jesenakova B, Babusikova E. Pulmonary manifestations of primary immunodeficiency disorders in children. Front Pediatr 2014;2:77. https://doi.org/10.3389/fped.2014.00077.
[7] Walter JE, Ayala IA, Milojevic D. Autoimmunity as a continuum in primary immunodeficiency. Curr Opin Pediatr 2019;31:851–62. https://doi.org/10.1097/MOP.0000000000000833.
[8] Orange JS, Seeborg FO, Boyle M, Scalchunes C, Hernandez-Trujillo V. Family Physician Perspectives on Primary Immunodeficiency Diseases. Front Med 2016;3. https://doi.org/10.3389/fmed.2016.00012.
[9] Hong J, Knutsen AP. Pulmonary Disease in Primary Immunodeficiency Disorders. Pediatric Allergy, Immunology, and Pulmonology 2013;26:57–68. https://doi.org/10.1089/ped.2013.0227.
[10] Lugo Reyes SO, Condino-Neto A, Stepensky P. Global perspectives on primary immune deficiency diseases. Stiehm’s Immune Deficiencies 2020:1129–42. https://doi.org/10.1016/B978-0-12-816768-7.00054-5.
[11] Salute M della. Esenzioni per malattie rare n.d. http://www.salute.gov.it/portale/esenzioni/dettaglioContenutiEsenzioni.jsp?lingua=italiano&id=1015&area=esenzioni&menu=vuoto (accessed August 15, 2020).
[12] Makita S, Hosoba R, Tobinai K. Safety considerations with targeted therapy drugs for B-cell non-Hodgkin lymphoma. Expert Opin Drug Saf 2020. https://doi.org/10.1080/14740338.2020.1802424.
[13] Leiding JW, Forbes LR. Mechanism-Based Precision Therapy for the Treatment of Primary Immunodeficiency and Primary Immunodysregulatory Diseases. The Journal of Allergy and Clinical Immunology: In Practice 2019;7:761–73. https://doi.org/10.1016/j.jaip.2018.12.017.
[14] Ochs HD, Hagin D. Primary immunodeficiency disorders: general classification, new molecular insights, and practical approach to diagnosis and treatment. Ann Allergy Asthma Immunol 2014;112:489–95. https://doi.org/10.1016/j.anai.2014.04.007.
[15] Tangye SG, Al-Herz W, Bousfiha A, Chatila T, Cunningham-Rundles C, Etzioni A, et al. Human Inborn Errors of Immunity: 2019 Update on the Classification from the International Union of Immunological Societies Expert Committee. Journal of Clinical Immunology 2020;40:24–64. https://doi.org/10.1007/s10875-019-00737-x.
[16] Shahin T, Aschenbrenner D, Cagdas D, Bal SK, Conde CD, Garncarz W, et al. Selective loss of function variants in <em>IL6ST</em> cause Hyper-IgE syndrome with distinct impairments of T-cell phenotype and function. Haematologica 2019;104:609–21. https://doi.org/10.3324/haematol.2018.194233.
[17] Spencer S, Köstel Bal S, Egner W, Lango Allen H, Raza SI, Ma CA, et al. Loss of the interleukin-6 receptor causes immunodeficiency, atopy, and abnormal inflammatory responses. Journal of Experimental Medicine 2019;216:1986–98. https://doi.org/10.1084/jem.20190344.
[18] Ma CS, Tangye SG. Flow Cytometric-Based Analysis of Defects in Lymphocyte Differentiation and Function Due to Inborn Errors of Immunity. Frontiers in Immunology 2019;10. https://doi.org/10.3389/fimmu.2019.02108.
[19] Fischer A, Rausell A. Primary immunodeficiencies suggest redundancy within the human immune system. Science Immunology 2016;1. https://doi.org/10.1126/sciimmunol.aah5861.
[20] Thrasher AJ, Williams DA. Evolving Gene Therapy in Primary Immunodeficiency. Mol Ther 2017;25:1132–41. https://doi.org/10.1016/j.ymthe.2017.03.018.
[21] Gathmann B, Goldacker S, Klima M, Belohradsky BH, Notheis G, Ehl S, et al. The German national registry for primary immunodeficiencies (PID). Clin Exp Immunol 2013;173:372–80. https://doi.org/10.1111/cei.12105.