20/10/2020
Le sindromi da iper-IgM
(Ultimo aggiornamento: 26/10/2020)
Le sindromi da iper-IgM (SIGM) costituiscono un gruppo di rare immunodeficienze primitive congenite ed ereditarie caratterizzata da [1]:
- Infezioni ricorrenti ad insorgenza precoce,
- Diminuzione estrema o assenza nel siero di IgG, IgE, IgA
- Livelli di IgM nel siero normali o aumentati
- Aumentato rischio di neoplasie e malattie autoimmuni.
Le cellule di alcune di queste sindrome mostrano aumentata sensibilità alle radiazioni in vitro [2]. Le infezioni croniche o ricorrenti, se non conotrollate, con la profilassi-terapia possono complicarsi con amiloidosi secondaria [3]. (Vedere "Amiloidosi - Una panoramica su epidemiologia, patogenesi, diagnosi e terapia ")
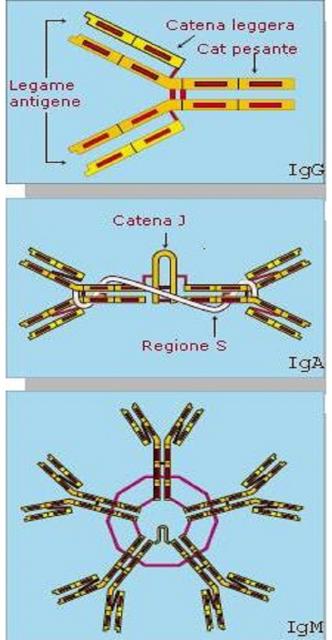
Si tratta di sindromi fenotipicamente e geneticamente eterogenee, conseguenti a difetti genetici che alterano i processi di “class switch recombination” (il cambio di classe o isotipo delle immunoglobuline”) e di ipermutazione somatica” necessari per la formazione di anticorpi di classe diversa dalle IgM, cioè di IgG, IgA, IgE [4]. Il cambio di isotipo immunoglobulinico e l’ipermutazione somatica avvengono nei centri germinativi dei follicoli secondari linfonodali e consentono la sintesi da parte dei linfociti B di anticorpi con maggiore affinità verso i patogeni [5]. La formazione di immunoglobuline non IgM è tipica della risposta immunitaria umorale secondaria, in assenza della quale, come avviene nelle sindromi da iper-IgM, si stabilisce uno stato di immunodeficienza per mancanza di immunoglobuline ad elevata affinità di legame per gli antigeni.
Come abbiamo detto le IgM possono essere normali o addirittura diminuite [6], per cui il termine di sindrome da iper-IgM può risultare fuorviante.Inoltre l'aumento delle IgM di per sè non causa grandi problemi, per cui sarebbe più corretto usare il termine di sindromi con iper-IgM.
Cenni sulla ricombinazione genica e l’ipermutazione somatica
Durante la prima fase del loro sviluppo (antigene indipendente) i linfociti B, in seguito alla ricombinazione V(D)J dei geni per la regione variabile, formano un unico gene per la regione variabile che viene poi assemblato con il gene dalle regione costante Cm , formando un unico gene per la catena pesante di classe M [7]. Un simile riarrangiamento si verifica a carico dei geni per la regione V delle catene leggere k o l. Le regioni costanti e variabile unendosi formano una molecola completa di classe IgM che è espressa sulla superficie dei linfociti B ed è anche secreta nel sangue periferico. La seconda fase di maturazione linfocitaria B si svolge nei linfonodi, dove i linfociti T, attivati dalle cellule dendritiche e dalle altre APC (Antigen Presenting Cells), stimolano i linfociti B a completare la loro maturazione e a produrre anticorpi di classe diversa dalle IgM.
I linfociti T, tramite il CD40L espresso sulla loro superficie, si legano al CD40 espresso sui linfociti B e sulle APC. Le interazioni CD40/CD40L attivano il CD40 che invia all’interno della cellula dei segnali indispensabili per la proliferazione dei linfociti B, per la formazione dei centri germinativi dei follicoli linfatici e per la formazione dei linfociti B memoria [8]. La stimolazione del CD40 attiva nelle cellule dendritiche il NfkB che induce l’espressione di geni che codificano per molecole costrimolatrici, come CD80 e CD86. Queste molecole sono richieste per un’efficace attivazione dei linfociti T e per la secrezione di citochine come IL-12 che promuovono la differenziazione dei linfociti T CD4+ naive in cellule Th1 [8].
Fra i geni attivati nei linfociti B che stanno attraversando i centri germinativi vi sono AID (Activation-induced deaminasi) ed UNG (Uracil-N glicosilasi), che introducono nella sequenza del DNA delle variazioni con una frequenza molto elevata. Questo processo di ipermutazione somatica consente di selezionare i linfociti B che producono anticorpi con la maggiore affinità verso i patogeni; questi stessi linfociti diverranno poi linfociti B memoria [8].
Sono anche riattivati i geni RAG-1 e RAG-2, che durante la fase antigene-indipendente dello sviluppo dei linfociti B rendono possibile la ricombinazione V(D)J, e altri geni che codificano per proteine appartenenti a sistemi enzimatici che intervengono nei processi di riparazione del DNA [7, 9, 10]. Questo secondo gruppo di geni opera la sostituzione del gene della regione costante m dell’IgM sostituendolo con uno di classe diversa (g,a,e); il nuovo segmento genico della regione costante è assemblato con l’originario gene V, cosicché la nuova immunoglobulina manterrà la stessa specificità antigenica della IgM ma avrà proprietà effettrici diverse da quest’ultima che le consentiranno di eliminare l’agente patogeno molto più rapidamente. (Vedere anche "Genetica e Struttura Del Recettore Dei Linfociti T (TCR) " e " Immunodeficienze primitive - Cenni sulla risposta immunitaria")
Classicamente, sono 5 geni i geni le cui mutazioni sono considerate come causa congenita della che causano la SIGM [11, 12]:
- Mutazioni del gene CD40L, localizzato sulla banda cromosomica Xp26, che codifica per il ligando di CD40, CD40L o CD154, responsabili della HIGM di tipo 1 a trasmissione recessiva legata al sesso o X-linked, XL-SIGM, HIGM-1 (OMIM#308230)
- Mutazioni della citidina deaminasi indotta dall'attivazione (AID o AICDA), espressa nei linfociti B e necessaria per la loro differenziazione terminale, o HIGM-2 (OMIM #605258), il cui locus genetico è stato mappato sul cromosoma 12p13
- Mutazioni del gene CD40, (HIGM-3, OMIM# 606843) il cui locus è mappato sul cromosoma 20q12-q13.2.
- Mutazioni di NEMO, nuclear factor kappaB essential modulator (NEMO o IKKg) o HIGM-4, OMIM#616884)
- Mutazioni del gene UNG, situato sul cromosoma 12q23-q24.1, che codifica per la uracil DNA glicosilasi, HIGM-5, OMIM#606186.
I cinque difetti genetici sopra ricordati comprendono però soltanto una parte dei casi. Analizzando il DNA dei pazienti con per-IgM ma senza un difetto identificabile, sono stati successivamente identificati mutazioni di altri geni che si manifestano con un fenotipo simile ai precedenti: PI3K-d [13], INO80 [14], MSH6 [15], PMS2 [16]. Tuttavia numerose altre sindromi congenite hanno spesso, ma non sempre, un aumento delle IgM e sono generalmente classificate in altre categorie di immunodeficienza primitiva: deficit di nuclear factor kappa B essential modulator (NEMO) e le sindromi da anomala riparazione del DNA, per esempio atassia telangiectasia (AT), la sindrome di Nijmegen, il deficit di DNA Ligasi 4 e il deficit di Cernunnos/XRCC4-like factor [17].
In conclusione, oltre alle mutazioni di CD40L, difetti di numerosi altri geni che controllano alcune vie di segnalazione dei B linfociti B e di attivazione dei linfociti T, la Class switch recombination delle immunoglobuline (Ig-CSR), l’ipermutazione somatica e la riparazione del DNA possono causare un fenotipo HIGM e cioè: CD40L, CD40, nuclear factor-kappa-B essential modulator (NEMO/IKKγ),inhibitor of kappa light chain gene enhancer in B cells alpha (IκBα), nuclear factor kappa-Bsubunit 1 (NKFB1), activation-induced cytidine deaminase (AICDA), uracil-DNA glycosylase(UNG), ataxia telangiectasia mutated (ATM), post meiotic segregation increased 2 (PMS2), MutSHomolog 6 (MSH6), MutS Homolog 2 (MSH2), INO80, Nibrin/Nijmegenbreakage syndrome 1 (NBS1/NBN), meiotic recombination 11-Like Protein A (MRE11),recombination activating gene 2 (RAG2), phosphatidylinositol 3-kinase catalytic delta (PIK3CD), phosphatidylinositol 3-kinase regulatory subunit 1 alpha (PIK3R1), tumor necrosis factor receptor superfamily member 13B (TACI/TNFRSF13B), inducible T-cell costimulator(ICOS), CD19, B cell-activating factor receptor (BAFF-R/TNFRSF13C), LPS Responsive Beige-Like Anchor Protein (LRBA), phospholipase C gamma-2 (PLCG2), Bruton tyrosine kinase (BTK) lymphocyte activation molecule-associated protein (SAP), DNA ligase 4 e Cernunnos [17].
L’eziopatogenesi e le manifestazioni cliniche sono diverse nelle varie sindromi da iper-IgM e devono essere conosciute per effettuare una diagnosi precoce , valutare la prognosi e scegliere la terapia più adatta nel singolo caso [18]. La maggioranza di questi disordini si manifesta tipicamente come un deficit dell’immunità umorale, ma la forma X-linked, la più frequente in assoluto, e quelle a trasmissione autosomica dominante si presentano come una immunodeficienza combinata[18, 19] dal momento che il deficit genetico specifico è presente nei linfociti T e B. (Vedere "Immunodeficienze primitive combinate - Classificazione " )
La terapia delle diverse forme di sindrome da iper-IgM si basa sulla profilassi e terapia delle infezioni con antibiotici, somministrazione periodica di immunoglobuline. Il trapianto di midollo può guarire definitivamente molti dei casi più gravi [20].
Riferimenti bibliografici
- Levy J, Espanol-Boren T, Thomas C, Fischer A, Tovo P, Bordigoni P, Resnick I, Fasth A, Baer M, Gomez L et al: Clinical spectrum of X-linked hyper-IgM syndrome. J Pediatr 1997, 131(1 Pt 1):47-54. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=9255191.
- Fekrvand S, Mozdarani H, Delavari S, Sohani M, Nazari F, Kiaee F, Bagheri Y, Azizi G, Hassanpour G, Mozdarani S et al: Evaluation of Radiation Sensitivity in Patients with Hyper IgM Syndrome. Immunol Invest 2020, 10.1080/08820139.2020.1779288:1-17. https://www.ncbi.nlm.nih.gov/pubmed/32584193.
- Delplanque M, Galicier L, Oziol E, Ducharme-Benard S, Oksenhendler E, Buob D, Grateau G, Boutboul D, Georgin-Lavialle S: AA amyloidosis secondary to primary immune deficiency: about 40 cases including 2 new French cases and a systematic literature review. J Allergy Clin Immunol Pract 2020, 10.1016/j.jaip.2020.09.023. https://www.ncbi.nlm.nih.gov/pubmed/33007500.
- Levitt D, Haber P, Rich K, Cooper MD: Hyper IgM immunodeficiency. A primary dysfunction of B lymphocyte isotype switching. J Clin Invest 1983, 72(5):1650-1657. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=6605368.
- Li Z, Woo CJ, Iglesias-Ussel MD, Ronai D, Scharff MD: The generation of antibody diversity through somatic hypermutation and class switch recombination. Genes Dev 2004, 18(1):1-11. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=14724175
- Heinold A, Hanebeck B, Daniel V, Heyder J, Tran TH, Dohler B, Greil J, Muller FM: Pitfalls of "hyper"-IgM syndrome: a new CD40 ligand mutation in the presence of low IgM levels. A case report and a critical review of the literature. Infection 2010, 38(6):491-496. https://www.ncbi.nlm.nih.gov/pubmed/20981468.
- Wang CL, Wabl M: DNA acrobats of the Ig class switch. J Immunol 2004, 172(10):5815-5821. file://C:%5CDocuments%20and%20Settings%5Cutente%5CDocumenti%5Cbiblio%5Cbibliografia%5CDNA%20Acrobats%20of%20the%20Ig.pdf.
- Cleary AM, Insel RA, Lewis DB: Disorders of lymphocyte function. In: Hematology: Basic Principles and practice. edn. Edited by Hoffman R, Benz EJ, Shattil S, Furie B, Cohen HJ, Silberstein L, McGlave P. Philadelphia: Elsevier; 2005: 831-855.
- Maizels N: Secret sharers in the immune system: a novel RNA editing activity links switch recombination and somatic hypermutation. Genome Biol 2000, 1(4):REVIEWS1025. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=11178252
- Bassing CH, Swat W, Alt FW: The mechanism and regulation of chromosomal V(D)J recombination. Cell 2002, 109 Suppl:S45-55. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=11983152
- Gulino AV, Notarangelo LD: Hyper IgM syndromes. Curr Opin Rheumatol 2003, 15(4):422-429. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=12819470
- Etzioni A, Ochs HD: The hyper IgM syndrome--an evolving story. Pediatr Res 2004, 56(4):519-525. https://www.ncbi.nlm.nih.gov/pubmed/15319456.
- Angulo I, Vadas O, Garcon F, Banham-Hall E, Plagnol V, Leahy TR, Baxendale H, Coulter T, Curtis J, Wu C et al: Phosphoinositide 3-kinase delta gene mutation predisposes to respiratory infection and airway damage. Science 2013, 342(6160):866-871. https://www.ncbi.nlm.nih.gov/pubmed/24136356.
- Kracker S, Di Virgilio M, Schwartzentruber J, Cuenin C, Forveille M, Deau MC, McBride KM, Majewski J, Gazumyan A, Seneviratne S et al: An inherited immunoglobulin class-switch recombination deficiency associated with a defect in the INO80 chromatin remodeling complex. J Allergy Clin Immunol 2015, 135(4):998-1007 e1006. https://www.ncbi.nlm.nih.gov/pubmed/25312759.
- Gardès P, Forveille M, Alyanakian M-A, Aucouturier P, Ilencikova D, Leroux D, Rahner N, Mazerolles F, Fischer A, Kracker S et al: Human MSH6 Deficiency Is Associated with Impaired Antibody Maturation. The Journal of Immunology 2012, 188(4):2023-2029. https://www.jimmunol.org/content/jimmunol/188/4/2023.full.pdf.
- Péron S, Metin A, Gardès P, Alyanakian M-A, Sheridan E, Kratz CP, Fischer A, Durandy A: Human PMS2 deficiency is associated with impaired immunoglobulin class switch recombination. Journal of Experimental Medicine 2008, 205(11):2465-2472. https://doi.org/10.1084/jem.20080789.
- Tangye SG, Al-Herz W, Bousfiha A, Chatila T, Cunningham-Rundles C, Etzioni A, Franco JL, Holland SM, Klein C, Morio T et al: Human Inborn Errors of Immunity: 2019 Update on the Classification from the International Union of Immunological Societies Expert Committee. J Clin Immunol 2020, 40(1):24-64. https://www.ncbi.nlm.nih.gov/pubmed/31953710.
- Yazdani R, Fekrvand S, Shahkarami S, Azizi G, Moazzami B, Abolhassani H, Aghamohammadi A: The hyper IgM syndromes: Epidemiology, pathogenesis, clinical manifestations, diagnosis and management. Clin Immunol 2019, 198:19-30. https://www.ncbi.nlm.nih.gov/pubmed/30439505.
- de la Morena MT: Clinical Phenotypes of Hyper-IgM Syndromes. J Allergy Clin Immunol Pract 2016, 4(6):1023-1036. https://www.ncbi.nlm.nih.gov/pubmed/27836054.
- Castagnoli R, Delmonte OM, Calzoni E, Notarangelo LD: Hematopoietic Stem Cell Transplantation in Primary Immunodeficiency Diseases: Current Status and Future Perspectives. Front Pediatr 2019, 7:295. https://www.ncbi.nlm.nih.gov/pubmed/31440487.