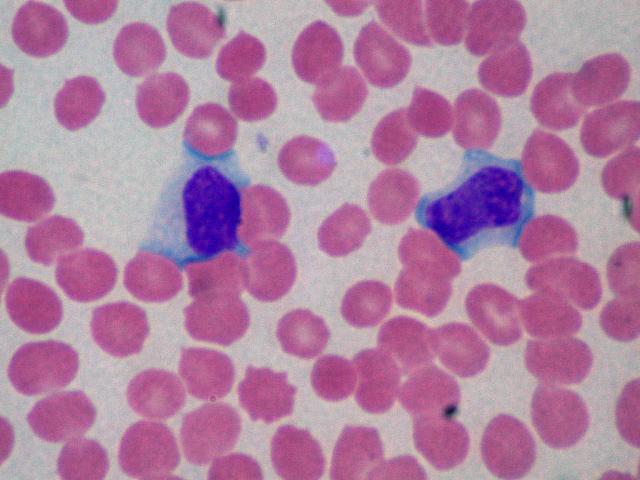
Linfociti atipici della mononucleosi infettiva
01/03/2020
Approccio al bambino con linfocitosi
(Ultimo aggiornamento: 29/08/2020)
Introduzione
I linfociti sono le principali cellule del sistema immunitario. Ne esistono diverse sottopopolazioni, non identificabili morfologicamente al microscopio ottico, ognuna delle quali è dotata di specifiche funzioni, che possono essere identificate con la citometria a flusso (Johansson et al. 2014). Questa metodica di laboratorio permette di distinguere facilmente e rapidamente anche una linfocitosi reattiva da una neoplastica, che è l’aspetto che più interessa il medico e il paziente. In soggetti con più di 12 anni, nella maggior parte dei laboratori si definisce linfocitosi un numero di linfociti superiore a 4.000/mL, ma alcuni considerano “normale” un limite superiore di 5000/mL (Vasu e Caligiuri 2016). Questi livelli sono più elevati nei neonati e nei bambini, che possono avere anche una linfocitosi assoluta superiore a 15.000 nelle prime settimane di vita e a 9.000/mL fino ai primi tre anni (Cordiano 2004b; Lanzkwosky, Lipton, e Fish 2016). Molto più frequente della linfocitosi assoluta è la linfocitosi relativa, cioè un aumento della sola percentuale di linfociti che si osserva in molti condizioni di neutropenia. Si tratta di un’alterazione che non è quasi mai espressione di una patologia primitiva linfocitaria.
I linfociti circolanti nel sangue periferico comprendono una popolazione eterogenea di linfociti T, di linfociti B e di cellule Natural Killer (NK), morfologicamente non sempre distinguibili. L’identificazione delle sottopopolazioni linfocitarie è notevolmente facilitata dalla citometria a flusso, la quale, usa anticorpi monoclonali fluorescenti diretti contro proteine di membrana relativamente specifiche per le varie linee linfocitarie. La metodica permette di distinguere agevolmente i diversi tipi di linfociti (Quantz et al. 1987a; Johansson et al. 2014). Ogni antigene di membrana è identificato in citometria con la sigla CD seguita da un numero arabico. Con queste metodiche le percentuali delle cellule linfoidi circolanti nel sangue periferico sono le seguenti: linfociti T (CD3+) 60-80%; linfociti B (CD20+) 10-20%; cellule NK (CD56+) 5-10% (Lanzkwosky, Lipton, e Fish 2016; Andrews et al. 2008). Le percentuali relative dei sottotipi principali di linfociti T del sangue sono linfociti T Helper/inducer (CD3+/CD4+) 60-70% e Linfociti T suppressor/citotossici (CD3+/CD8+) 30-40%, con un rapporto CD4+/CD8+ di circa 2:1 (Roquiz, Al Diffalha, e Kini 2016). (Vedere " Immunodeficienze primitive - Cenni sulla risposta immunitaria")
La conta linfocitaria assoluta è oggi fornita di routine con l’emocromo in tutti i laboratori di analisi cliniche. Poiché le cause di linfocitosi assoluta sono numerose e spesso poco evidenti, spesso sono necessari esami dispendiosi sia in termini economici sia in termini di tempo per arrivare ad una diagnosi conclusiva (George 2012).
Definizioni e valori normali
L'intervallo di riferimento "normale" (2 deviazioni standard sopra o sotto la media) per i leucociti negli adulti varia da 4400 a 11.000 cellule/mL nella maggioranza dei laboratori. La percentuale dei linfociti generalmente costituisce il 10-33% dei globuli bianchi sangue periferico.
La percentuale relativa dei linfociti non è un indice attendibile per valutare l'aumento di linfociti dal momento che può essere aumentata anche in caso di riduzione di una o più delle altre popolazioni leucocitarie (linfocitosi relativa). Importante e, quindi, riferirsi il numero assoluto dei linfociti quando si voglia valutare da un punto di vista quantitativo il numero reale di queste cellule nella conta differenziale leucocitaria (Cordiano 2004b).
Il numero assoluto dei linfociti può essere calcolato con la seguente formula :
Numero assoluto di linfociti/mL = [(numero di leucociti/mL) × percentuale dei linfociti]/100.
Pertanto, se in numero totale di leucociti è 10.500/mL e la percentuale dei linfociti è del 32%, il numero assoluto di linfociti sarà = 3360/mL (10.500 × 32 = 336.000/100 = 3360).
Una linfocitosi assoluta è definita generalmente nell'adulto con un numero assoluto di linfociti >4000/mL. Per linfocitopenia si intende una riduzione del numero assoluto dei linfociti< 1500/mL.
Queste definizioni sono valide in generale, ma aumenti di alcune sottopopolazioni linfocitarie possono assumere un ruolo particolarmente significativo anche con un numero di linfociti assoluti normale. Le proliferazioni clonali croniche di linfociti sono rare nei bambini. Linfocitosi solitamente secondaria a infezioni o infiammazioni croniche con associata neutropenia.
Patogenesi della linfocitosi
Il processo di differenziazione dei linfociti a partire dai precursori midollari e timici, della loro maturazione e della loro differenziazione in cellule effettrici del sistema immunitario è molto complesso e avviene in diverse fasi durante tutta la vita dell’individuo nel midollo emopoietico e negli organi linfoidi secondari (milza, linfonodi, timo ecc.) (Roquiz, Al Diffalha, e Kini 2016). Una linfocitosi può prodursi per aumentata produzione, per alterata distribuzione nel sangue o, infine, per aumento della durata vita media (riduzione dell’apoptosi) di una o più sottopopolazioni linfocitarie. In alcuni più meccanismi intervengono nel produrre l’aumento dei linfociti.
La linfocitosi può essere innanzitutto distinta in due grandi categorie:
1) linfocitosi reattiva o policlonale;
2) Linfocitosi primitiva, quasi sempre monoclonale.
Linfocitosi primitiva
La linfocitosi primitiva si riferisce a pazienti con una diagnosi accertata di malattia linfoproliferativa acuta o cronica. Esempi di questi disordini sono la leucemia linfatica cronica, la leucemia linfoblastica acuta, la leucemia dei grandi linfociti granulati. La linfocitosi primitiva è quasi sempre monoclonale, ma è necessario ricordare che monoclonalità non è sinonimo di malignità, in quanto risposte monoclonali transitorie possono essere osservate normalmente nel corso delle risposte immunitarie ad agenti infettivi o in soggetti con malattie non neoplastiche (Bashford-Rogers et al. 2019). Inoltre nella linfocitosi primitiva è compresa anche la linfocitosi policlonale B persistente (Vasu e Caligiuri 2016).
La clonalità dei linfociti è oggi facilmente dimostrabile nel caso dei linfociti B con la citometria a flusso, meno nei linfociti T e nelle cellule NK(Verschoor, Kohli, e Balion 2018; Johansson et al. 2014). La presenza di una popolazione monoclonale comporta un’alterazione del rapporto fra catena leggera kappa (k) e lambda (l ) delle immunoglobuline di superficie. Dal momento che ogni linfocito produce molecole di Ig con catena leggera k o l, mai entrambe contemporaneamente (fenomeno noto anche come restrizione k /l) è evidente come un eccesso di linfociti k o l comporterà rispettivamente un aumento o una diminuzione del rapporto k /l (Rawstron, Hillmen, e Houlston 2004).
Altri metodi per stabilire la clonalità, ognuno con una sensibilità diversa, sono (Rockman 1997; McCarthy et al. 2015; Bashford-Rogers et al. 2019; Vasu e Caligiuri 2016; Grange et al. 2017):
- La dimostrazione di un cariotipo anormale come la traslocazione t(12; 21), caratteristica di una malattia leucemia linfoblastica acuta pediatrica, o la t(14;18), tipica del linfoma follicolare, ma che raramente è presente in altre MLPC ;
- Il riarrangiamento clonale dei geni variabili della catena pesante delle immunoglobuline (IgVH), che si osserva frequentemente nelle leucemie/linfomi delle cellule B;
- Il riarrangiamento clonale del recettore per le cellule T (TCR), che si osserva in diverse leucemie o linfomi a cellule T. Per definizione, le neoplasie maligne delle cellule NK non hanno riarrangiamenti clonali dei geni per le immunoglobuline o di quelli per il recettore per le cellule T. Tuttavia, come nel caso delle leucemie aggressive delle cellule NK, hanno spesso geni del virus di Epstein-Barr clonale integrato nel DNA cellulare( Kanegane et al. 1998) e/o anomalie cromosomiche clonali.
Linfomi e leucemie
Soprattutto nelle fasi precoci delle malattie linfoproliferative dei linfociti B, T, o NK, nel bambino possono comparire cellule che non sono morfologicamente molto diverse dai linfociti normali policlonali o da quelli reattivi. Le cellule linfoidi neoplastiche che compaiono nel sangue periferico hanno spesso una morfologia caratteristica, tanto che ci si può indirizzare rapidamente verso la diagnosi di una delle malattie linfoproliferative (Hamad e Mangla 2019). L’esame del midollo, l'immunofenotipo, gli studi del riarrangiamento genico, la citogenetica sono poi necessari per differenziare la leucemia dalla linfocitosi benigna e per stabilire il fenotipo delle cellule leucemiche ai fini della terapia (Quantz et al. 1987b; Cordiano 2004b; George 2012).
Nei bambini e nei giovani adulti, la presenza di linfociti attivati e di una linfocitosi non spiegata può essere dovuta a linfoblasti pre-B circolanti, suggestivi di una diagnosi di leucemia linfoblastica acuta (LLA). I linfoblasti della leucemia linfoblastica acuta possono avere una morfologia molto diversa, variabile da cellule praticamente prive di citoplasma, passando per cellule con abbondante citoplasma debolmente basofilo fino all’aspetto di linfoblasti tipo "cellule di Burkitt.” con citoplasma intensamente basofilo e spiccata vacuolizzazione.
Nel sangue periferico dei bambini sono frequenti linfociti normali che possono essere scambiati per forme immature e linfoblasti da un osservatore poco esperto. Nel midollo dei bambini più piccoli possono essere presenti gli "ematogoni", precursori normali dei linfociti B in via di maturazione di aspetto immaturo , a volte difficilmente distinguibili con la sola morfologia dai linfoblasti. Gli ematogoni possono arrivare a costituire oltre il 6% dei linfociti periferici nel 60% degli infanti con meno di un mese di età (Brady, Atwater, e Lowell 1999), mentre nell'adulto costituiscono meno dell'1% dei leucociti periferici valutati con metodi di flussocitometria che permettono di identificare correttamente questo raro tipo di cellule (Sevilla et al. 2010). Il sospetto di LLA deve aumentare se l’emocromo del bambino dimostra oltre alla linfocitosi con cellule anomale anche anemia e/o piastrinopenia.
Altre neoplasie linfoidi possono presentare, durante il decorso della malattia, un numero variabile di cellule anomale nel sangue periferico il cui aspetto può essere abbastanza suggestivo per indirizzare verso la diagnosi. Nei bambini le leucemie croniche e i linfomi indolenti sono meno frequenti rispetto agli adulti.
Linfocitosi monoclonale dei linfociti B
La diffusione della citometria a flusso oggi ha permesso di distinguere facilmente la linfocitosi monoclonale, che è spesso, ma non sempre, di natura neoplastica - da quella policlonale o reattiva permettendo di ottenere rapidamente la diagnosi definitiva (Quantz et al. 1987b; Rockman 1997; Blumberg e Schooley 1985a; Johansson et al. 2014). La LLC-B, la più frequente fra le linfocitosi primitive, ha un fenotipo caratteristico che la distingue dalle altre malattie linfoproliferative: cellule CD5+/CD19+/CD79b-/CD20+/CD23+/sIg con restrizione k /l (Hallek et al. 2008; Johansson et al. 2014.) La LLC-B origina virtualmente in tutti i casi da uno stato preneoplastico, la linfocitosi monoclonale a cellule B (MBL, monoclonal B-cell lymphocytosis), simile a quello della MGUS che, in molti casi, precede il mieloma multiplo (Maitre e Troussard 2019). (vedere anche " Linfocitosi monoclonale dei linfociti B – Definizioni") La diagnosi di MBL può effettuarsi in un paziente con linfocitosi monoclonale assoluta< 5000/microlitro/mL (il livello soglia, cioè, necessario per una diagnosi di LLC-B) in assenza di sintomi e segni di malattie autoimmuni, infezioni, altre malattie linfoproliferative o di altre cause di linfocitosi nota (Maitre e Troussard 2019). Da notare che una popolazione monoclonale di linfociti B può essere presente anche in alcuni soggetti con un numero assoluto di linfociti< 4000/ mL, che è il livello soglia accettato della maggior parte dei laboratori per una diagnosi di linfocitosi. Spesso questi pazienti hanno un familiare affetto da LLC-B o altra malattia linfoproliferativa oppure, malattie autoimmuni (Goldin e Slager 2007; Sellick, Catovsky, e Houlston 2006). Cellule B con il fenotipo delle cellule LLC-B sono presenti anche in soggetti sani con un conta linfocitaria normale (Rawstron, Hillmen, e Houlston 2004). La MBL aumenta il rischio di LLC-B o di altre neoplasie linfoidi B anche nei familiari di primo grado del probando e, anche se la maggioranza dei casi di MBL non progredisce mai verso una malattia maligna del sistema linfatico, è associata ad una prognosi peggiore con riduzione della vita media (Maitre e Troussard 2019; Angelillo et al. 2018; Strati e Shanafelt 2015; Angelillo et al. 2018).
La diagnosi di MBL è posta dopo aver escluso in base alla storia, all'esame obiettivo, e agli esami radiologici che il paziente non abbia un linfoma, una malattia autoimmune oppure un'infezione (Cordiano 2004b). È raccomandato che i soggetti con MBL eseguano una visita annuale per monitorare questo disordine linfoproliferativo la cui diagnosi precoce può permettere di identificare i casi a maggior rischio da seguire con controlli più ravvicinati (Marti et al. 2005; Maitre e Troussard 2019). (Vedere anche "Linfocitosi monoclonale dei linfociti B – Epidemiologia, fattori di rischio, storia naturale e rischio di evoluzione in leucemia ")
Malattia linfoproliferativa dei grandi linfociti granulati
Il sospetto di un’espansione monoclonale dei grandi linfociti granulati sorge in presenza di una linfocitosi atipica con caratterizzata da >2.000/m L di grandi linfociti con nucleo eccentrico, citoplasma abbondante e pallido contenente numerosi granuli azurofili (Liu e Loughran 2011).
Da un punto di vista fenotipico queste cellule, che sono solitamente monoclonali, possono avere il fenotipo di cellule T CD3+/CD 8+/CD4- oppure di cellule NK CD3-/CD56+ (Loughran 1993).I pazienti con questa forma di linfocitosi possono essere asintomatici oppure avere febbre, infezioni, artrite, neutropenia, anemia e o piastrinopenia (Barilà et al. 2019; Semenzato et al. 1997; Liu e Loughran 2011). La dimostrazione della clonalità del TCR è necessaria per differenziare le forme neoplastiche da quelle reattive della espansione degli LGL(Semenzato et al. 1997; Vasu e Caligiuri 2016),
Linfocitosi congenita delle cellule B
Una linfocitosi si osserva in rare famiglie i cui membri sono portatori di mutazioni di CARD11 (caspase recruitment domain containing protein 11), una proteina necessaria per l’attivazione di nuclear factor-kappa B nei linfociti B e T. I bambini si presentano con splenomegalia, infezioni ricorrenti, anergia e linfocitosi policlonale dei linfociti B che può evolvere a LLC-B o altra malattia linfoproliferativa entro la quarta decade di vita(Snow et al. 2012; Shields et al. 2020). (Vedere anche "Deficit di CARD11 ")
Morfologia linfocitaria
Lo studio della morfologia linfocitaria è di grande aiuto nella diagnosi differenziale delle linfocitosi, soprattutto nelle mani di un osservatore esperto che riesce a distinguere nella maggior parte una linfocitosi reattiva da un caso di linfocitosi primitiva e, in alcuni casi, ad arrivare alla diagnosi rapidamente specie se conosce l’età e la clinica del paziente (Vasu e Caligiuri 2016; Hamad e Mangla 2019; Ravandi e O’Brien 2005).
Linfocitosi reattiva
Per linfocitosi secondaria o reattiva intendiamo un aumento del numero assoluto dei linfociti secondario ad una risposta fisiologica o fisiopatologica a infezioni, tossine, citochine, traumi o a fattori sconosciuti (Vasu e Caligiuri 2016). La linfocitosi reattiva si riferisce ad una linfocitosi in genere policlonale in un paziente senza storia di malattie ematologiche, con una patologia nota per poter essere associata con linfocitosi e nel quale ci si attende che il numero di linfociti si normalizzi in meno di 2 mesi dalla guarigione della patologia associata. Esempi possono essere le infezioni virali e la pertosse.
In generale, una linfocitosi reattiva è sospettata in presenza nel sangue periferico di uno dei due tipi seguenti di linfociti: un aumento assoluto di piccoli linfociti d’aspetto maturo, come si osserva nella pertosse (figura 1) e nella linfocitosi infettiva; oppure un aumento assoluto dei linfociti e presenza di elementi di dimensioni più grandi (linfociti attivati o virociti) dai contorni irregolari, con citoplasma abbondante e basofilo, nucleo grande e irregolare contenente a volte nucleoli, cromatina a volte fine che ricorda quella dei linfoblasti (Hamad e Mangla 2019; Vasu e Caligiuri 2016). La mononucleosi infettiva acuta durante la seconda o la terza settimana di malattia si caratterizza per un marcato aumento di questi linfociti, spesso denominati linfociti atipici o attivati. La caratteristica principale dei “linfociti attivati” è il polimorfismo, cioè la notevole variabilità dell’aspetto degli elementi cellulari, che si contrappone ad una relativa uniformità dell’aspetto delle cellule nelle linfocitosi neoplastiche. Un quadro monomorfo orienterà l’operatore verso una diagnosi diversa. I linfociti “attivati” possono essere osservati anche in altre malattie virali elencate nella tabella 1. Le cause principali di linfocitosi reattiva sono le infezioni virali frequentemente osservate nei bambini e nei giovani adulti (tabella 1)
Mononucleosi infettiva
L'infezione da virus di Epstein-Barr (EBV) è la principale causa di mononucleosi infettiva (Hamad e Mangla 2019; Vasu e Caligiuri 2016), malattia caratterizzata dalla comparsa, soprattutto nella seconda o terza settimana, di numerosi linfociti atipici reattivi i quali possono essere ancora presenti dopo due mesi. Nonostante il virus infetti i linfociti B, la linfocitosi reattiva è causata dall'aumento assoluto dei linfociti T CD3+/CD8+ suppressor che hanno attività contro i linfociti B EBV+ (Hudnall et al. 2003). Il rapporto CD4/CD8, normalmente attorno a 1,8-2, può scendere a 0,49 o anche meno (Blumberg e Schooley 1985b). Nella diagnosi differenziale possono aiutare la lieve epatite d’accompagnamento ed il rash cutaneo, che caratteristicamente, compare nei pazienti che assumano antibiotici o, meno frequentemente, altri farmaci (Paily 2000; McCloskey e Massa 1997)
Sindrome mononucleosica EBV negativa
Un quadro clinico simile a quello della mononucleosi infettiva EBV+ può essere causato da diversi agenti infettivi, i più importanti dei quali sono citomegalovirus (CMV) e HIV-1 (Hurt e Tammaro 2007)
Nel primo periodo della malattia CMV causa un aumento dei linfociti T CD8+, con riduzione del rapporto CD4+/CD8+, ma durante la convalescenza ricomincia la risalita de linfociti T CD4+ ed il rapporto si normalizza (Carney et al. 1981).
Durante l'infezione primaria da HIV-1 i pazienti possono presentarsi con una sindrome mononucleosica nei casi con infezione contemporanea da CMV (Zambello et al. 1993; Hurt e Tammaro 2007). Nell'infezione primaria da HIV-1 i linfociti del sangue periferico hanno un fenotipo CD3+/CD8+, cioè di tipo suppressor-citotossico (Cooper et al. 1985).
Altre infezioni virali sono state occasionalmente considerate come causa della sindrome mononucleosica, inclusi il virus Human Herpes Virus 6 (HHV-6), l’adenovirus tipo 1(Hamad e Mangla 2019; Vasu e Caligiuri 2016) e 2 e il virus HTLV-1 che provoca una linfocitosi prevalente dei linfociti T CD4+ con un rapporto CD4/CD8 4-5 : 1(Ehrlich et al. 1988).
I pazienti affetti da varicella, influenza, parotite, rosolia e altre infezioni virali solitamente hanno una linfocitosi reattiva (Cordiano 2004b; Blumberg e Schooley 1985a). L’analisi delle sottopolazioni linfocitarie ha evidenziato frequentemente un’inversione del rapporto CD4+/CD8+ per riduzione dei linfociti T CD4+ (Blumberg e Schooley 1985b).
Linfocitosi infettiva acuta
Nei bambini numerosi virus, compresi enterovirus e poliovirus, sono stati associati con una condizione benigna chiamata linfocitosi infettiva acuta (Cassuto et al. 1977; Vasu e Caligiuri 2016). I pazienti hanno una leucocitosi periferica variante da 20.000 a 100.000/m L con il 60-70% di linfociti piccoli maturi di tipo T. La malattia è spesso accompagnata da una lieve eosinofilia e si risolve in 4-10 settimane dalla diagnosi che, solitamente, è posta casualmente. La maggioranza dei pazienti è, infatti, asintomatica, sebbene siano stati riportati casi decorsi con febbre, segni e sintomi respiratori, diarrea, dolore addominale; raramente sono stati segnalati casi con meningite, encefalite o rash morbilliforme. Spesso è scambiata per una leucemia o per una mononucleosi infettiva(Vasu e Caligiuri 2016).
Pertosse
Le infezioni batteriche acute raramente producono linfocitosi, tranne la pertosse che è causata dalla Bordetella pertussis. In uno studio con pochi casi la conta linfocitaria assoluta variava da 6.500 a 54.800/mL con una media di 23.000/mL(Kubic, Kubic, e Brunning 1991). I linfociti sono in prevalenza piccoli linfociti con nucleo inciso: il 20% di essi è di tipo B il 50% di linfociti T. Quindi la linfocitosi della pertosse è dovuta all'espansione di linfociti normali (Kubic, Kubic, e Brunning 1991). L'aumento di questa popolazione sembra essere il risultato del blocco della normale migrazione dei linfociti dal sangue ai linfonodi. La tossina purificata della pertosse blocca una G-proteina richiesta per la trasmissione dei segnali calcio dipendente nei linfociti, e interferisce con il normale processo di circolo dei linfociti dal sangue ai linfonodi (Vasu e Caligiuri 2016). L'iniezione endovenosa della tossina produce una transitoria linfocitosi nelle scimmie, con aumento soprattutto dei linfociti CD 4+ rispetto a quelli CD8+ (Hinds et al. 1996; Vasu e Caligiuri 2016). La linfocitosi manca in una malattia quasi identica causata dalla Bordetella parapertussis, anche se quest'ultima produce la stessa tossina della pertosse (Heininger et al. 1994).
Malattia da graffio di gatto
La malattia da graffio di gatto è causata dalla Bartonella henselae ed è caratterizzata da una linfoadenopatia solitaria con segni sistemici e sintomi d’infiammazione cronica. Grandi linfociti atipici possono essere osservati nello striscio periferico, solitamente con una lieve eosinofilia, monocitosi, anemia e piastrinopenia.
Infezioni -batteriche croniche
Altre infezioni croniche come infezioni da Rickettsie, tubercolosi, brucellosi e sifilide spesso causano una linfocitosi cronica
Toxoplasmosi
La toxoplasmosi solitamente produce un’infezione asintomatica o una linfoadenopatia nei pazienti immunocompetenti (cosiddetta toxoplasmosi linfoadenopatica); può causare tuttavia un'infezione disseminata nei pazienti immunocompromessi. La linfoadenopatia è solitamente multipla e in un terzo dei casi è stata associata con epatosplenomegalia. Come ricordato in precedenza, il quadro ematologico può simulare quello della mononucleosi infettiva, con linfociti numerosi reattivi grandi e atipici; eosinofilia si osserva nel 10-20% dei casi (Remington 1974; Vasu e Caligiuri 2016).
Una causa rara di linfocitosi è la Babesiosi, una zoonosi trasmessa con la puntura di zecche che può essere causata da varie specie di Babesia (B. microbi, B. divergens). Il parassita causa anemia emolitica penetrando nei globuli rossi, simulando clinicamente la malaria. I parassiti sono facilmente dimostrati all’interno degli eritrociti nello striscio periferico.
Cause non infettive di linfocitosi reattiva
Reazione da ipersensibilità (da farmaci, malattia da siero acuta) e lo stress sono le cause più frequenti della linfocitosi reattiva non infettiva (Vasu e Caligiuri 2016; Hamad e Mangla 2019).
Reazioni allergiche
Le reazioni da ipersensibilità ritardata a punture di insetto, specialmente zanzare, sono spesso associate ad aumento dei LGL e linfoadenopatia satellite (Roh et al. 2010). Le reazioni idiosincrasiche a farmaci tipicamente insorgono dopo 2-8 settimane dall’inizio dell’assunzione del farmaco. I sulfamidici possono causare una sindrome mononucleosica (Vasu e Caligiuri 2016). La linfocitosi si osserva nel 30-70% dei casi della sindrome nota come Drug reaction with eosinophilia and systemic symptoms (DRESS)(Hamad e Mangla 2019).
Linfocitosi da stress
Una linfocitosi atipica transitoria con numero di linfociti variabile da 4.000 a a 13.000/mL, spesso seguita da una neutrofilia, è stata osservata in soggetti con emergenze cardiache, traumi o stato epilettico (Teggatz, Parkin, e Peterson 1987). La mortalità era del 50% e la linfocitosi era considerata come la conseguenza dello stress o della somministrazione di adrenalina. La presenza di linfocitosi dopo un trauma sembra conferire una prognosi peggiore (Pinkerton et al. 1989). Anche in pazienti adulti con crisi occlusive dell'anemia falciforme, traumi o altre malattie acute mediche è possibile osservare un aumento assoluto dei linfociti (Groom et al. 1990; Teggatz, Parkin, e Peterson 1987). Questi dati sembrano simili a quelli osservati dopo somministrazione parenterale di catecolamine: dopo un rapido aumento dei linfociti segue una loro riduzione con aumento dei neutrofili (Vasu e Caligiuri 2016). La linfocitosi da stress è un esempio di linfocitosi da alterato ricircolo dei linfociti in quanto riguarda tutte le sottopopolazioni linfocitarie (Vasu e Caligiuri 2016).
Valutazione
Un bambino con linfocitosi può rappresentare un caso da risolvere urgentemente, per esempio se nello striscio periferico è segnalata la presenza di blasti leucemici o se le condizioni cliniche del paziente mostrano un rapido deterioramento. In questo caso il sospetto di una leucemia o di linfoma aggressivo è giustificato e il bambino deve essere inviato in ospedale. Se il bambino è sintomatico e non ha una precedente diagnosi di emopatia, soprattutto se le sue condizioni cliniche non sono stabili, e l'emocromo evidenzia anomalie preoccupanti, per esempio la presenza di forme immature ed una linfocitosi elevata, pe >20.000-30.000 cellule/microlitro, è opportuno ripetere l'emocromo con urgenza.
Nella maggior parte dei casi la linfocitosi è però asintomatica e di entità modesta e può essere valutata con più calma in ambulatorio.
L'anamnesi, l'esame obiettivo è la valutazione dello striscio periferico, assieme agli altri parametri dell'emocromo rappresentano il cardine della valutazione del bambino con linfocitosi (Hamad e Mangla 2019). Se il piccolo paziente si presenta con sintomi quali febbre, calo ponderale, linfoadenopatia, splenomegalia, petecchie o altre emorragie cutanee e delle mucose, con piastrinopenia e/o anemia all'emocromo, è necessario escludere che si tratti di un linfoma/leucemia aggressivo, per esempio una leucemia linfoblastica acuta o un linfoma di Burkitt in un bambino, mentre in un paziente adulto il quadro clinico potrebbe essere sostenuta da un linfoma ad istotipo aggressivo.
Nella pratica clinica odierna, la linfocitosi si incontra più frequentemente nel corso di una valutazione di un esame emocromocitometrico richiesto di routine in soggetti sani e asintomatici oppure nel corso del monitoraggio delle patologie più svariate.
Se il quadro clinico non preoccupa e la linfocitosi è isolata (assenza di grave anemia e/o piastrinopenia) e già nota, o può essere documentata con un precedente emocromo, la valutazione del paziente può procedere con più calma. L'anamnesi attenta potrà chiarire l'esistenza di una delle cause note di linfocitosi, per esempio l'assunzione di farmaci, la splenectomia o altre malattie croniche autoimmuni, infiammatorie, infettive o tumorali associate alla linfocitosi (Hamad e Mangla 2019; Vasu e Caligiuri 2016).
I sintomi e i segni clinici prima ricordati, come pure le anomalie dell'emocromo, possono essere osservati anche in soggetti con mononucleosi infettiva o altre malattie infettive virali elencate nella tabella. In questo caso la presenza di linfociti atipici o attivati, assieme agli opportuni test sierologici può indirizzare verso la corretta diagnosi.
L'esame obiettivo deve essere indirizzato alla ricerca di segni di infezioni, infiammazioni, o neoplasie con particolare attenzione alla febbre, linfoadenopatia, epatosplenomegalia, tumefazioni delle articolazioni, dolore addominale, masse mediastiniche, emorragie cutanee e delle mucose orali, ipertrofia gengivale. In caso di grave anemia si potrà osservare pallore delle congiuntive e delle mucose e il paziente potrà presentare di dispnea, ipotensione, tachicardia. L'anemia questi pazienti può essere del tipo più vario: anemia normocitica delle malattie croniche nei pazienti con malattie infiammatorie o infettive croniche; emolitica; aplastica in corso di timoma o di malattia dei grandi linfociti granulati. La positività degli indici di flogosi (VES, proteina C reattiva), l'assetto marziale (sideremia, transferrina, ferritina), la bilirubinemia possono suggerire la causa della linfocitosi svelando, per esempio, una patologia infiammatoria cronica in caso di aumento della VES e/o della PCR in un bambino asintomatico.
Un ruolo importante nella ricerca della corretta diagnosi della linfocitosi hanno naturalmente gli esami sierologici per le molte malattie infettive croniche o autoimmuni che possono causare la linfocitosi.
Valutazione della clonalità
Il passo successivo è la distinzione fra linfocitosi clonale e policlonale, distinzione che non può essere effettuata con le metodiche utilizzate per la valutazione iniziale del paziente, cioè anamnesi, esame obiettivo, emocromo, indici di flogosi e gli altri test di laboratorio finora considerati.
La citometria a flusso è la metodica più semplice per la valutazione della clonalità della linfocitosi (Bain e Haferlach 2011). Tuttavia, la presenza di una popolazione monoclonale o di piccole popolazioni oligoclonali non è sinonimo di neoplasia, poiché numerose malattie infiammatorie o infettive causano linfocitosi con espansione oligoclonale dei linfociti caratterizzata dalla presenza di 2-5 piccoli cloni linfocitari (Chen e Moller 2014; Dustin e Charles 2012).
Un clone di ridotte dimensioni nell'ambito di una popolazione più numerosa di linfociti policlonale è caratteristico della linfocitosi monoclonale delle cellule B, una condizione che può progredire a LLC-B conclamata e che può osservarsi anche in familiari di pazienti con LLC-B nota (Maitre e Troussard 2019). In questi casi la corretta diagnosi potrà avvenire soltanto nel corso del follow-up con la ripetizione dell'emocromo e, eventualmente, della flussocitometria e degli altri test specializzati di cui parleremo fra poco. Non tutti pazienti con linfocitosi richiedono una flussocitometria, che va riservata ai pazienti che richiedono una precisazione diagnostica urgente, per esempio il sospetto di una leucemia linfoblastica acuta o di altro linfoma aggressivo, oppure in caso di linfocitosi di cospicua entità di primo riscontro (Bain e Haferlach 2011).
Negli altri casi meno urgenti, e sono la maggioranza, è opportuno ripetere dopo 4 settimane l'emocromo è richiedere la valutazione della clonalità e dell'immunofenotipo nei seguenti casi: un numero assoluto di linfociti > 5000/microlitro; pazienti con citopenie periferiche, epatosplenomegalia, linfoadenopatia; pazienti con presenza di blasti o linfociti atipici nello striscio periferico; aumento significativo del numero di linfociti rispetto all'emocromo precedente.
Tecniche di genetica molecolare sono necessarie per dimostrare la monoclonalità dei geni per le immunoglobuline di superficie dei linfociti B o dei geni del TCR nei linfociti T, mentre la dimostrazione della clonalità delle cellule NK è molto più complessa(Bain e Haferlach 2011).
La clonalità dei linfociti può essere evidenziata anche con l'analisi cromosomica mediante cariotipo convenzionale o ibridazione fluorescenza in situ (FISH) che possono evidenziare anomalie cromosomiche specifiche per alcune patologie linfoproliferative maligne, per esempio la traslocazione t(11;14) tipica del linfoma mantellare o la traslocazione Bcr/abl tipica della leucemia linfoblastica Ph+.
La clonallità linfocitaria può essere dimostrata anche sul materiale bioptico o citologico (linfonodi, tessuti vari) prelevato per la diagnosi istologica di linfoma necessaria quando l’immunofenotipo su sangue periferico o midollare deponga per una malattia linfoproliferativa diversa dalla LLC.B.
Conclusioni
La linfocitosi è un problema che il pediatra deve affrontare frequentemente nella sua pratica clinica quotidiana. Nella maggioranza dei casi si tratta di condizioni reattive o benigne, la cui diagnosi è relativamente agevole in base all’emocromo e a pochi altri esami disponibili presso i normali laboratori. In caso di persistenza della linfocitosi e/o della presenza di anomalie dell’emocromo concomitanti (anemia, piastrinopenia, blasti) è indicata una rapida consulenza ematologica.
Riferimenti bibliografici
Andrews, Jared M., Dan L. Cruser, Jerome B. Myers, Colby A. Fernelius, Mitchel T. Holm, e Dale L. Waldner. 2008. «Using Peripheral Smear Review, Age and Absolute Lymphocyte Count as Predictors of Abnormal Peripheral Blood Lymphocytoses Diagnosed by Flow Cytometry». Leukemia & Lymphoma 49 (9): 1731–37. https://doi.org/10.1080/10428190802251787.
Angelillo, Piera, Antonella Capasso, Paolo Ghia, e Lydia Scarfò. 2018. «Monoclonal B-Cell Lymphocytosis: Does the Elderly Patient Need a Specialistic Approach?» European Journal of Internal Medicine 58: 2–6. https://doi.org/10.1016/j.ejim.2018.09.006.
Bain, Barbara J, e Torsten Haferlach. 2011. «Laboratory diagnosis of haematological neoplasms». In Postgraduate Haematology, a cura di A Victor Hoffbrand, Daniel Catovsky, Edward GD Tuddenham, e Anthony R. Green, 6th ed. Wiley-Blackwell.
Barilà, Gregorio, Giulia Calabretto, Antonella Teramo, Cristina Vicenzetto, Vanessa Rebecca Gasparini, Gianpietro Semenzato, e Renato Zambello. 2019. «T Cell Large Granular Lymphocyte Leukemia and Chronic NK Lymphocytosis». Best Practice & Research. Clinical Haematology 32 (3): 207–16. https://doi.org/10.1016/j.beha.2019.06.006.
Barton, A. D. 1997. «T-Cell Lymphocytosis Associated with Lymphocyte-Rich Thymoma». Cancer 80 (8): 1409–17. https://doi.org/10.1002/(sici)1097-0142(19971015)80:8<1409::aid-cncr7>3.0.co;2-9.
Bashford-Rogers, R. J. M., L. Bergamaschi, E. F. McKinney, D. C. Pombal, F. Mescia, J. C. Lee, D. C. Thomas, et al. 2019. «Analysis of the B Cell Receptor Repertoire in Six Immune-Mediated Diseases». Nature 574 (7776): 122–26. https://doi.org/10.1038/s41586-019-1595-3.
Bernard, C., H. Frih, F. Pasquet, S. Kerever, Y. Jamilloux, F. Tronc, B. Guibert, et al. 2016. «Thymoma Associated with Autoimmune Diseases: 85 Cases and Literature Review». Autoimmunity Reviews 15 (1): 82–92. https://doi.org/10.1016/j.autrev.2015.09.005.
Blumberg, R. S., e R. T. Schooley. 1985a. «Lymphocyte Markers and Infectious Diseases». Seminars in Hematology 22 (2): 81–114.
———. 1985b. «Lymphocyte Markers and Infectious Diseases». Seminars in Hematology 22 (2): 81–114.
Brady, K. A., S. K. Atwater, e C. A. Lowell. 1999. «Flow Cytometric Detection of CD10 (CALLA) on Peripheral Blood B Lymphocytes of Neonates». British Journal of Haematology 107 (4): 712–15. https://doi.org/10.1046/j.1365-2141.1999.01775.x.
Burton, Andrew G., Dori L. Borjesson, e William Vernau. 2014. «Thymoma-Associated Lymphocytosis in a Dog». Veterinary Clinical Pathology 43 (4): 584–88. https://doi.org/10.1111/vcp.12196.
Byrd, John C., Susan O’Brien, e Danelle F. James. 2013. «Ibrutinib in Relapsed Chronic Lymphocytic Leukemia». The New England Journal of Medicine 369 (13): 1278–79. https://doi.org/10.1056/NEJMc1309710.
Carney, W. P., R. H. Rubin, R. A. Hoffman, W. P. Hansen, K. Healey, e M. S. Hirsch. 1981. «Analysis of T Lymphocyte Subsets in Cytomegalovirus Mononucleosis». Journal of Immunology (Baltimore, Md.: 1950) 126 (6): 2114–16.
Cassuto, J. P., M. Schneider, M. Bourg, A. Bertrand, e R. Mariani. 1977. «Acute Infectious Lymphocytosis as a T-Cell Lymphoproliferative Syndrome». British Medical Journal 2 (6098): 1331–32. https://doi.org/10.1136/bmj.2.6098.1331-a.
Chen, Edward S., e David R. Moller. 2014. «Etiologic Role of Infectious Agents». Seminars in Respiratory and Critical Care Medicine 35 (3): 285–95. https://doi.org/10.1055/s-0034-1376859.
Cooper, D. A., J. Gold, P. Maclean, B. Donovan, R. Finlayson, T. G. Barnes, H. M. Michelmore, P. Brooke, e R. Penny. 1985. «Acute AIDS Retrovirus Infection. Definition of a Clinical Illness Associated with Seroconversion». Lancet (London, England) 1 (8428): 537–40. https://doi.org/10.1016/s0140-6736(85)91205-x.
Cordiano, Vincenzo. 2004a. «Leucemia linfatica cronica». In La Medicina di laboratorio nella pratica medica., 416–24. Selecta Medica.
———. 2004b. «Linfocitosi». In La Medicina di laboratorio nella pratica medica., 363–64. Selecta Medica.
Cranney, A., S. Markman, B. Lach, e J. Karsh. 1997. «Polymyositis in a Patient with Thymoma and T Cell Lymphocytosis». The Journal of Rheumatology 24 (7): 1413–16.
Delage, R., L. Jacques, M. Massinga-Loembe, J. Poulin, D. Bilodeau, C. Mignault, P. F. Leblond, e A. Darveau. 2001. «Persistent Polyclonal B-Cell Lymphocytosis: Further Evidence for a Genetic Disorder Associated with B-Cell Abnormalities». British Journal of Haematology 114 (3): 666–70. https://doi.org/10.1046/j.1365-2141.2001.02975.x.
Deplano, Simona, Elizabet Nadal‐Melsió, e Barbara J. Bain. 2014. «Persistent Polyclonal B Lymphocytosis». American Journal of Hematology 89 (2): 224–224. https://doi.org/10.1002/ajh.23630.
Doll, D. C., R. J. Landreneau, e A. F. List. 1991. «Malignant Thymoma Associated with Peripheral T-Cell Lymphocytosis». Medical and Pediatric Oncology 19 (6): 496–98. https://doi.org/10.1002/mpo.2950190609.
Dustin, Lynn B., e Edgar D. Charles. 2012. «Primary, Post-Primary and Non-Specific Immunoglobulin M Responses in HCV Infection». Antiviral Therapy 17 (7 Pt B): 1449–52. https://doi.org/10.3851/IMP2222.
Ehrlich, G. D., T. Han, R. Bettigole, S. A. Merl, B. Lehr, R. H. Tomar, e B. J. Poiesz. 1988. «Human T-Lymphotropic Virus Type I-Associated Benign Transient Immature T-Cell Lymphocytosis». American Journal of Hematology 27 (1): 49–55. https://doi.org/10.1002/ajh.2830270112.
Garcia-Suarez, J., A. Prieto, E. Reyes, K. Arribalzaga, M. A. Perez-Machado, M. Lopez-Rubio, L. Manzano, e M. Alvarez-Mon. 1995. «Persistent Lymphocytosis of Natural Killer Cells in Autoimmune Thrombocytopenic Purpura (ATP) Patients after Splenectomy». British Journal of Haematology 89 (3): 653–55. https://doi.org/10.1111/j.1365-2141.1995.tb08382.x.
George, Tracy I. 2012. «Malignant or Benign Leukocytosis». Hematology 2012 (1): 475–84. https://doi.org/10.1182/asheducation.V2012.1.475.3798515.
Goldin, Lynn R., e Susan L. Slager. 2007. «Familial CLL: Genes and Environment». Hematology 2007 (1): 339–45. https://doi.org/10.1182/asheducation-2007.1.339.
Grange, Béatrice, Evelyne Callet-Bauchu, Gilles Salles, e Pierre Sujobert. 2017. «Advances in the Role of Cytogenetic Analysis in the Molecular Diagnosis of B-Cell Lymphomas». Expert Review of Molecular Diagnostics 17 (6): 623–32. https://doi.org/10.1080/14737159.2017.1327811.
Groom, D. A., L. A. Kunkel, R. K. Brynes, J. W. Parker, C. S. Johnson, e D. Endres. 1990. «Transient Stress Lymphocytosis during Crisis of Sickle Cell Anemia and Emergency Trauma and Medical Conditions. An Immunophenotyping Study». Archives of Pathology & Laboratory Medicine 114 (6): 570–76.
Hallek, Michael, Bruce D. Cheson, Daniel Catovsky, Federico Caligaris-Cappio, Guillaume Dighiero, Hartmut Döhner, Peter Hillmen, et al. 2008. «Guidelines for the Diagnosis and Treatment of Chronic Lymphocytic Leukemia: A Report from the International Workshop on Chronic Lymphocytic Leukemia Updating the National Cancer Institute-Working Group 1996 Guidelines». Blood 111 (12): 5446–56. https://doi.org/10.1182/blood-2007-06-093906.
Hamad, Hussein, e Ankit Mangla. 2019. «Lymphocytosis». In StatPearls. Treasure Island (FL): StatPearls Publishing. http://www.ncbi.nlm.nih.gov/books/NBK549819/.
Han, Xiang Y., Pei Lin, Hesham M. Amin, e Alessandra Ferrajoli. 2005. «Postsplenectomy Cytomegaloviral Mononucleosis: Marked Lymphocytosis, TCRgamma Gene Rearrangements, and Impaired IgM Response». American Journal of Clinical Pathology 123 (4): 612–17. https://doi.org/10.1309/HLBB-K8V0-A6T8-BYV8.
Heininger, U., K. Stehr, S. Schmitt-Grohé, C. Lorenz, R. Rost, P. D. Christenson, M. Uberall, e J. D. Cherry. 1994. «Clinical Characteristics of Illness Caused by Bordetella Parapertussis Compared with Illness Caused by Bordetella Pertussis». The Pediatric Infectious Disease Journal 13 (4): 306–9. https://doi.org/10.1097/00006454-199404000-00011.
Himmelmann, A., O. Gautschi, M. Nawrath, U. Bolliger, J. Fehr, e R. A. Stahel. 2001. «Persistent Polyclonal B-Cell Lymphocytosis Is an Expansion of Functional IgD(+)CD27(+) Memory B Cells». British Journal of Haematology 114 (2): 400–405. https://doi.org/10.1046/j.1365-2141.2001.02938.x.
Hinds, P. W., C. Yin, M. S. Salvato, e C. D. Pauza. 1996. «Pertussis Toxin Induces Lymphocytosis in Rhesus Macaques». Journal of Medical Primatology 25 (6): 375–81. https://doi.org/10.1111/j.1600-0684.1996.tb00032.x.
Hudnall, S. David, Jyoti Patel, Hanna Schwab, e José Martinez. 2003. «Comparative Immunophenotypic Features of EBV-Positive and EBV-Negative Atypical Lymphocytosis». Cytometry. Part B, Clinical Cytometry 55 (1): 22–28. https://doi.org/10.1002/cyto.b.10043.
Hurt, Christopher, e Dominick Tammaro. 2007. «Diagnostic Evaluation of Mononucleosis-like Illnesses». The American Journal of Medicine 120 (10): 911.e1-8. https://doi.org/10.1016/j.amjmed.2006.12.011.
Johansson, Ulrika, David Bloxham, Stephen Couzens, Jennifer Jesson, Ricardo Morilla, Wendy Erber, Marion Macey, e British Committee for Standards in Haematology. 2014. «Guidelines on the Use of Multicolour Flow Cytometry in the Diagnosis of Haematological Neoplasms. British Committee for Standards in Haematology». British Journal of Haematology 165 (4): 455–88. https://doi.org/10.1111/bjh.12789.
Juneja, S., E. Januszewicz, M. Wolf, e I. Cooper. 1995. «Post-Splenectomy Lymphocytosis». Clinical and Laboratory Haematology 17 (4): 335–37.
Kaito, K., H. Otsubo, Y. Ogasawara, T. Shimada, K. Kasama, Y. Yahagi, O. Asai, N. Usui, e M. Kobayashi. 2000. «Severe Aplastic Anemia Associated with Chronic Natural Killer Cell Lymphocytosis». International Journal of Hematology 72 (4): 463–65.
Kanegane, H., A. Yachie, T. Miyawaki, e G. Tosato. 1998. «EBV-NK Cells Interactions and Lymphoproliferative Disorders». Leukemia & Lymphoma 29 (5–6): 491–98. https://doi.org/10.3109/10428199809050908.
Kreutzman, A., K. Ladell, C. Koechel, E. Gostick, M. Ekblom, L. Stenke, T. Melo, et al. 2011. «Expansion of Highly Differentiated CD8+ T-Cells or NK-Cells in Patients Treated with Dasatinib Is Associated with Cytomegalovirus Reactivation». Leukemia 25 (10): 1587–97. https://doi.org/10.1038/leu.2011.135.
Kreutzman, Anna, Vesa Juvonen, Veli Kairisto, Marja Ekblom, Leif Stenke, Ruth Seggewiss, Kimmo Porkka, e Satu Mustjoki. 2010. «Mono/Oligoclonal T and NK Cells Are Common in Chronic Myeloid Leukemia Patients at Diagnosis and Expand during Dasatinib Therapy». Blood 116 (5): 772–82. https://doi.org/10.1182/blood-2009-12-256800.
Kubic, V. L., P. T. Kubic, e R. D. Brunning. 1991. «The Morphologic and Immunophenotypic Assessment of the Lymphocytosis Accompanying Bordetella Pertussis Infection». American Journal of Clinical Pathology 95 (6): 809–15. https://doi.org/10.1093/ajcp/95.6.809.
Lanzkwosky, Phylip, Jeffrey H Lipton, e Jonathan D Fish. 2016. «Hematological Reference Values». In LANZKOWSKY’S MANUAL OF PEDIATRIC HEMATOLOGY AND ONCOLOGY, Sixth, 719.
Lesesve, Jean Francois, e Xavier Troussard. 2011. «Persistent Polyclonal B-Cell Lymphocytosis». Blood 118 (25): 6485. https://doi.org/10.1182/blood-2011-01-331082.
Lishner, M., M. Ravid, J. Shapira, J. Radnay, A. Amiel, V. Leytin, C. Shapiro, e A. Klein. 1994. «Delta-T-Lymphocytosis in a Patient with Thymoma». Cancer 74 (11): 2924–29. https://doi.org/10.1002/1097-0142(19941201)74:11<2924::aid-cncr2820741106>3.0.co;2-q.
Liu, Xin, e Thomas P. Loughran. 2011. «The Spectrum of Large Granular Lymphocyte Leukemia and Felty’s Syndrome». Current Opinion in Hematology 18 (4): 254–59. https://doi.org/10.1097/MOH.0b013e32834760fb.
Loembé, M. M., J. Lamoureux, N. Deslauriers, A. Darveau, e R. Delage. 2001. «Lack of CD40-Dependent B-Cell Proliferation in B Lymphocytes Isolated from Patients with Persistent Polyclonal B-Cell Lymphocytosis». British Journal of Haematology 113 (3): 699–705. https://doi.org/10.1046/j.1365-2141.2001.02806.x.
Loughran, T. P. 1993. «Clonal Diseases of Large Granular Lymphocytes». Blood 82 (1): 1–14.
Macintyre, E. A., e D. C. Linch. 1988. «Lymphocytosis: Is It Leukaemia and When to Treat». Postgraduate Medical Journal 64 (747): 42–47. https://doi.org/10.1136/pgmj.64.747.42.
Maitre, Elsa, e Xavier Troussard. 2019. «Monoclonal B-Cell Lymphocytosis». Best Practice & Research. Clinical Haematology 32 (3): 229–38. https://doi.org/10.1016/j.beha.2019.06.002.
Marti, Gerald E., Andy C. Rawstron, Paolo Ghia, Peter Hillmen, Richard S. Houlston, Neil Kay, Thérèse A. Schleinitz, Neil Caporaso, e International Familial CLL Consortium. 2005. «Diagnostic Criteria for Monoclonal B-Cell Lymphocytosis». British Journal of Haematology 130 (3): 325–32. https://doi.org/10.1111/j.1365-2141.2005.05550.x.
McCarthy, Brian A., Sophia Yancopoulos, Mike Tipping, Xiao-Jie Yan, Xue Ping Wang, Fiona Bennett, Wentian Li, et al. 2015. «A Seven-Gene Expression Panel Distinguishing Clonal Expansions of Pre-Leukemic and Chronic Lymphocytic Leukemia B Cells from Normal B Lymphocytes». Immunologic Research 63 (1–3): 90–100. https://doi.org/10.1007/s12026-015-8688-3.
McCloskey, G. L., e M. C. Massa. 1997. «Cephalexin Rash in Infectious Mononucleosis». Cutis 59 (5): 251–54.
Montserrat, E., N. Viñolas, J. C. Reverter, e C. Rozman. 1988. «Natural History of Chronic Lymphocytic Leukemia: On the Progression and Progression and Prognosis of Early Clinical Stages». Nouvelle Revue Francaise D’hematologie 30 (5–6): 359–61.
Paily, R. 2000. «Quinolone Drug Rash in a Patient with Infectious Mononucleosis». The Journal of Dermatology 27 (6): 405–6. https://doi.org/10.1111/j.1346-8138.2000.tb02192.x.
Paydas, Semra. 2014. «Dasatinib, Large Granular Lymphocytosis, and Pleural Effusion: Useful or Adverse Effect?» Critical Reviews in Oncology/Hematology 89 (2): 242–47. https://doi.org/10.1016/j.critrevonc.2013.10.005.
Pinkerton, P. H., B. A. McLellan, M. C. Quantz, e J. B. Robinson. 1989. «Acute Lymphocytosis after Trauma--Early Recognition of the High-Risk Patient?» The Journal of Trauma 29 (6): 749–51. https://doi.org/10.1097/00005373-198906000-00009.
Qiu, Zhi-Yuan, Wei Xu, e Jian-Yong Li. 2014. «Large Granular Lymphocytosis during Dasatinib Therapy». Cancer Biology & Therapy 15 (3): 247–55. https://doi.org/10.4161/cbt.27310.
Quantz, M. C., J. B. Robinson, V. Sachs, e P. H. Pinkerton. 1987a. «Lymphocyte Surface Marker Studies in the Diagnosis of Unexplained Lymphocytosis». CMAJ: Canadian Medical Association Journal = Journal de l’Association Medicale Canadienne 136 (8): 835–38.
Quantz, M C, J B Robinson, V Sachs, e P H Pinkerton. 1987b. «Lymphocyte surface marker studies in the diagnosis of unexplained lymphocytosis.» CMAJ: Canadian Medical Association Journal 136 (8): 835–38. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1492089/.
Ravandi, Farhad, e Susan O’Brien. 2005. «Chronic Lymphoid Leukemias Other than Chronic Lymphocytic Leukemia: Diagnosis and Treatment». Mayo Clinic Proceedings 80 (12): 1660–74. https://doi.org/10.4065/80.12.1660.
Rawstron, Andy, Peter Hillmen, e Richard Houlston. 2004. «Clonal Lymphocytes in Persons without Known Chronic Lymphocytic Leukemia (CLL): Implications of Recent Findings in Family Members of CLL Patients». Seminars in Hematology 41 (3): 192–200. https://doi.org/10.1053/j.seminhematol.2004.05.001.
Remington, J. S. 1974. «Toxoplasmosis in the Adult». Bulletin of the New York Academy of Medicine 50 (2): 211–27.
Rockman, S. P. 1997. «Determination of Clonality in Patients Who Present with Diagnostic Dilemmas: A Laboratory Experience and Review of the Literature». Leukemia 11 (6): 852–62. https://doi.org/10.1038/sj.leu.2400678.
Roh, Eui Jung, Eun Hee Chung, Young Pyo Chang, Na Hye Myoung, Young Koo Jee, Min Seo, e Jin Han Kang. 2010. «A Case of Hypersensitivity to Mosquito Bite Associated with Epstein-Barr Viral Infection and Natural Killer Cell Lymphocytosis». Journal of Korean Medical Science 25 (2): 321–23. https://doi.org/10.3346/jkms.2010.25.2.321.
Roquiz, Woodline, Sameer Al Diffalha, e Ameet R Kini. 2016. «Leukocyte Development, Kinetics, and Functions». In - Clinical principles and practice, a cura di Elaine M Kehoane, Larry J Smith, e Jeanine Walenga, 5th ed., 149–66. Elsevier.
Scorsetti, Marta, Francesco Leo, Annalisa Trama, Rolando D’Angelillo, Danila Serpico, Marianna Macerelli, Paolo Zucali, Gemma Gatta, e Marina Chiara Garassino. 2016. «Thymoma and Thymic Carcinomas». Critical Reviews in Oncology/Hematology 99 (marzo): 332–50. https://doi.org/10.1016/j.critrevonc.2016.01.012.
Sellick, Gabrielle S., Daniel Catovsky, e Richard S. Houlston. 2006. «Familial Chronic Lymphocytic Leukemia». Seminars in Oncology 33 (2): 195–201. https://doi.org/10.1053/j.seminoncol.2006.01.013.
Semenzato, G., R. Zambello, G. Starkebaum, K. Oshimi, e T. P. Loughran. 1997. «The Lymphoproliferative Disease of Granular Lymphocytes: Updated Criteria for Diagnosis». Blood 89 (1): 256–60.
Sevilla, Deborah W., Adriana I. Colovai, Foxwell N. Emmons, Govind Bhagat, e Bachir Alobeid. 2010. «Hematogones: A Review and Update». Leukemia & Lymphoma 51 (1): 10–19. https://doi.org/10.3109/10428190903370346.
Shields, Adrian M., Bradly M. Bauman, Chantal E. Hargreaves, Andrew J. Pollard, Andrew L. Snow, e Smita Y. Patel. 2020. «A Novel, Heterozygous Three Base-Pair Deletion in CARD11 Results in B Cell Expansion with NF-ΚB and T Cell Anergy Disease». Journal of Clinical Immunology, gennaio. https://doi.org/10.1007/s10875-019-00729-x.
Snow, Andrew L., Wenming Xiao, Jeffrey R. Stinson, Wei Lu, Benjamin Chaigne-Delalande, Lixin Zheng, Stefania Pittaluga, et al. 2012. «Congenital B Cell Lymphocytosis Explained by Novel Germline CARD11 Mutations». The Journal of Experimental Medicine 209 (12): 2247–61. https://doi.org/10.1084/jem.20120831.
Strati, Paolo, e Tait D. Shanafelt. 2015. «Monoclonal B-Cell Lymphocytosis and Early-Stage Chronic Lymphocytic Leukemia: Diagnosis, Natural History, and Risk Stratification». Blood 126 (4): 454–62. https://doi.org/10.1182/blood-2015-02-585059.
Teggatz, J. R., J. Parkin, e L. Peterson. 1987. «Transient Atypical Lymphocytosis in Patients with Emergency Medical Conditions». Archives of Pathology & Laboratory Medicine 111 (8): 712–14.
Templeton, Arnoud J., Mairéad G. McNamara, Boštjan Šeruga, Francisco E. Vera-Badillo, Priya Aneja, Alberto Ocaña, Raya Leibowitz-Amit, et al. 2014. «Prognostic Role of Neutrophil-to-Lymphocyte Ratio in Solid Tumors: A Systematic Review and Meta-Analysis». Journal of the National Cancer Institute 106 (6): dju124. https://doi.org/10.1093/jnci/dju124.
Vasu, Sumithira, e Michael A Caligiuri. 2016. «Lymphocytosis and lymphocytopenia». In Williams Hematology, a cura di K Kaushansky, Marshall A. Lichtman, J. T. Prchal, Olliver Press, Linda J Burns, e Michael A Caligiuri, 9th ed., 1199–1210. McGraw-Hill Education.
Verschoor, Chris P., Vikas Kohli, e Cynthia Balion. 2018. «A Comprehensive Assessment of Immunophenotyping Performed in Cryopreserved Peripheral Whole Blood». Cytometry Part B: Clinical Cytometry 94 (5): 818–26. https://doi.org/10.1002/cyto.b.21526.
Victor Hoffbrand, A., e Terry J. Hamblin. 2007. «Is “Leukemia” an Appropriate Label for All Patients Who Meet the Diagnostic Criteria of Chronic Lymphocytic Leukemia?» Leukemia Research 31 (3): 273–75. https://doi.org/10.1016/j.leukres.2006.07.006.
Voelxen, Nadine, Claudia Wehr, Sylvia Gutenberger, Baerbel Keller, Miriam Erlacher, Cecilia Dominguez-Conde, Daniela Bertele, et al. 2016. «B-Cell Signaling in Persistent Polyclonal B Lymphocytosis (PPBL)». Immunology and Cell Biology 94 (9): 830–37. https://doi.org/10.1038/icb.2016.46.
Woyach, Jennifer A., Kelly Smucker, Lisa L. Smith, Arletta Lozanski, Yiming Zhong, Amy S. Ruppert, David Lucas, et al. 2014. «Prolonged Lymphocytosis during Ibrutinib Therapy Is Associated with Distinct Molecular Characteristics and Does Not Indicate a Suboptimal Response to Therapy». Blood 123 (12): 1810–17. https://doi.org/10.1182/blood-2013-09-527853.
Zambello, R., L. Trentin, C. Agostini, P. Francia di Celle, E. Francavilla, A. Barelli, A. Cerutti, F. Siviero, R. Foà, e G. Semenzato. 1993. «Persistent Polyclonal Lymphocytosis in Human Immunodeficiency Virus-1-Infected Patients». Blood 81 (11): 3015–21.
|
Infezioni virali |
|
Mononucleosi infettiva (EBV +) |
|
Sindromi mononucleosiche EBV- (CMV, HHV-6, HHV-8 Adenovirus 12, HIV-1 ) |
|
Epatite acuta |
|
Rosolia, Morbillo, Varicella zoster, Influenza, Parotite |
|
HTLV-1 |
|
Linfocitosi infettiva acuta (Coxackie virus B12, adenovirus, enterovirus, altri?) |
|
Batteriche |
|
Pertosse, brucellosi, tubercolosi, m. da graffio di gatto, sifilide, listeriosi, leptospirosi |
|
Sindrome da tossina stafilococcica |
|
Lebbra |
|
Protozoarie |
|
Toxoplasmosi, Babesiosi, malaria, leishmaniosi,giardiasi |
|
Nematodi |
|
Strongiloidosi |
|
Farmaci |
|
Reazioni da ipersensibilità a farmaci |
|
Malattia da siero |
|
Stress |
|
Traumi |
|
Interventi chirurgici maggiori |
|
Collasso cardiovascolare: Insufficienza cardiaca acuta, infarto del miocardio, shock settico |
|
Stato di male epilettico |
|
Crisi occlusive dell’ anemia falciforme |
|
Punture d’insetto |
|
Fumo di sigarette |
|
Malattie autoimmuni |
|
Artrite reumatoide |
|
Sarcoidosi |
|
Granulomatosi di Wegener |
|
Dermatomiosite |
|
Vasculiti sistemiche |
|
Neoplasie |
|
Timoma |
|
Linfomi leucemizzati, LLC-B e sue varianti (L. prolinfocitica, LLC-T, L. dei grandi linfociti granulati, L. a cellule capellute) |
|
Tumori solidi |
|
Iposplensimo |
|
Endocrinopatie |
|
M.di Addison |
|
Ipertiroidismo |
|
Linfocitosi monoclonale delle celle B |
|
LInfocitosi policlonale peristente |
|
Linfocitosi delle cellule B congenita da mutazioni di CARD11 |